The actual function prediction of functionally unknown proteins in S. pneumoniae utilized the yeast-2-hybrid method that was supplemented with a bacterial meta-interactome. This bacterial meta-interactome allowed for the quality of the predictions to increase as compared to just using the yeast-2-hybrid method by itself, as described in previous experiments.
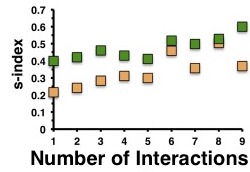
Wuchty et al. also wanted to look at the degree of accuracy of these predictions and how it related to the number of interaction partners of a given protein. The Simpson s-index was calculated for each newly characterized protein to assess the probability of a single protein function dominating for a given protein. The s-index ranges from 0 to 1 with 1 being a single function dominating for a protein while 0 is that all functions share an equal probability of being the function of a given protein. As expected, the accuracy of the functional prediction increased with the number of added protein interactions for both the original protein network and the protein network with the added bacterial meta-interactome, as the s-index’s grew towards one. The largest differences seen between the original and the added network was when there was a low amount of interaction partners within S. pneumoniae for a given protein.
Given that the bacterial meta-interactome supplementation has a positive impact on the quality of function prediction for proteins, Wuchty et al then set out to characterize 342 previously unknown or poorly characterized proteins in S. pneumoniae using this method. Using a false discovery rate of <0.05, 299 of the 342 proteins were able to be characterized with over 60% of the proteins having functions relating to transcription or translation.