Pyruvate ferredoxin oxidoreductase (PFOR) is an enzyme that converts pyruvate to Acetyl-CoA (known as pyruvate oxidation) in anaerobic bacteria. Specifically, PFOR has a cofactor in its active site, an adjunct helper molecule, called thiamine pyrophosphate (TPP). The pyruvate binds to TPP and the TPP catalyzes the conversion of pyruvate to acetyl CoA. To reiterate, TPP is a companion to PFOR which directly converts pyruvate to acetyl-CoA. PFOR also regenerates NAD, an electron carrier that is essential for ATP production.
Recall that pyruvate oxidation is one of the key steps in converting glucose to ATP to provide energy for the bacterium. If the energy production of an anaerobic bacteria were to be prevented, then the lack of ATP would ultimately cause the bacteria to die. In regards to pyruvate oxidation in anaerobic bacteria, a drug that could block pyruvate from binding to PFOR could potentially stop ATP production. Nitazoxanide (NTZ) and amixicile are such drugs that apparently blocks pyruvate from binding to TPP in PFOR.
Nitazoxanide (NTZ) is a drug that was observed in previous studies to deactivate TPP by abstracting a proton from TPP, thus preventing pyruvate conversion to Acetyl-CoA. Initially, NTZ was advocated as a potential all-encompassing drug, meaning that it would be able to target a variety of anaerobic bacteria. However, later studies found that since NTZ is required in large amounts to be effective, it may have harmful side effects. Kennedy et al. attempted to modify the molecular structure of NTZ to find a more effective derivative (a modified molecule) - Kennedy et al. found that amixicile was a potentially effective derivative of NTZ. Amixcile was deemed potentially effective because unlike NTZ, amixicile is effective in small amounts, as described in the Introduction.
Procedure
|
“In silico” is a term used for computer-generated. A docking simulation software is a tool that allows for the manipulation of 3D molecules of interest virtually. There are many different types of docking simulation software used to determine the structure, function, and interaction of molecules of interest. This experiment utilized the molecular operating environment (MOE) docking simulation software. MOE is a type of docking simulation software that specifically allows for the manipulation of drug molecules and predicts the manner of interaction between a drug molecule and the target molecule (that the drug binds to). For Kennedy et al’s study, the interaction of each NTZ and amixicile binding to the active site of PFOR were executed via MOE and observed. To reiterate, molecular operating environment is a specific type of docking simulation software that allows for the virtual modification of a drug molecule and implements the predicted interaction between a drug molecule and the target molecule (that the drug binds to).
The Triangle match algorithm is a specific procedure within the molecular operating environment (MOE) that implements organic chemistry reactions to allow for the simulation for the modification of the drug molecule of interest and interaction of a drug molecule and the target molecule (that the drug binds to). |
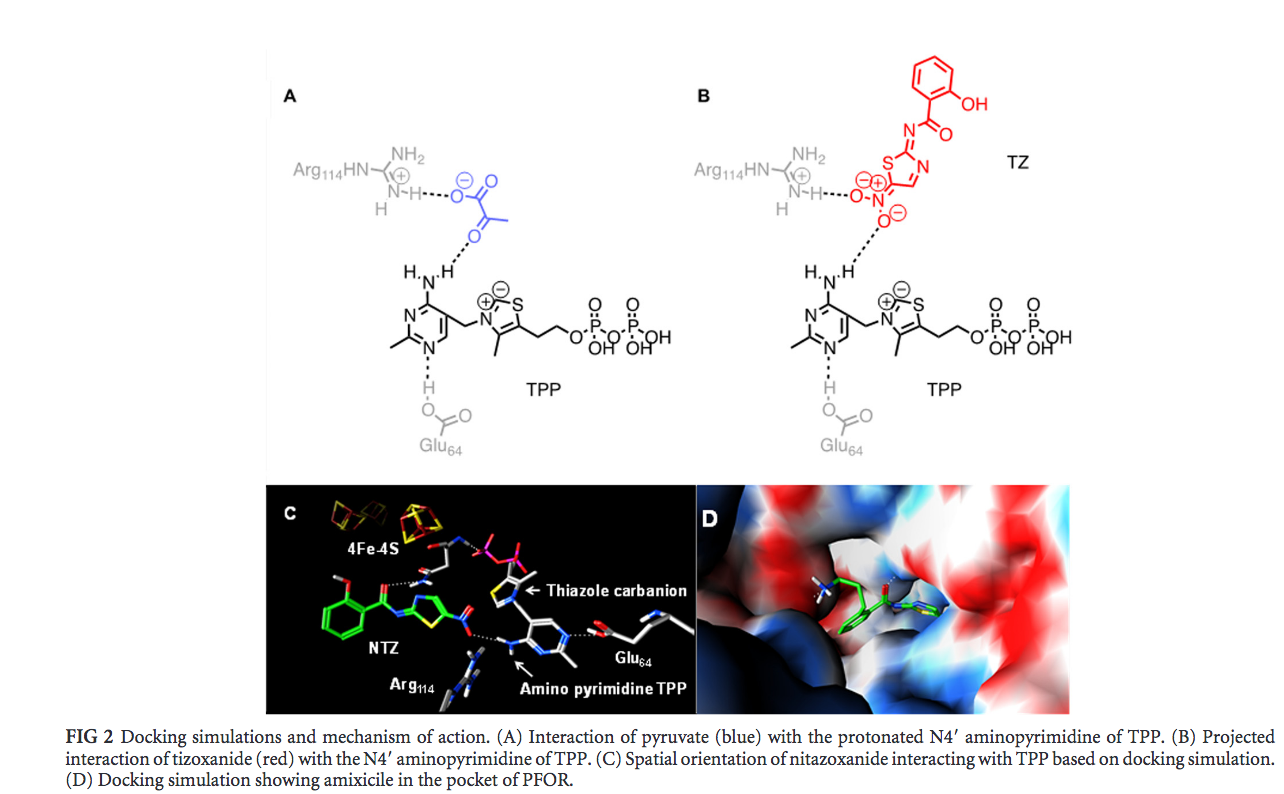
Fig. 2:Preview of mechanisms beind Molecular Operating Environment
|
In Kennedy et al’s study, Triangle match algorithm was implemented in the molecular operating environment (MOE) to dock amixicile and NTZ each in the active site of PFOR to compare their interactions with PFOR. Different orientations of binding were determined by the amount of possible free energy each orientation had. The “amount of possible free energy” is a fancy way to articulate the measurement of binding affinity between the drug molecule of interest and the target molecule (that the drug binds to) - in this case, the amount of possible free energy for each amixicile and NTZ were measured. The processes specifically used to determine the amount of possible free energy were the Merck molecular force field and London dG scoring function were specifically used to estimate and score the free energy of each possible orientation. The Merck molecular force field is a process within the Triangle match algorithm procedure which uses a specified parameter (particularly MM3 force field) to make organic chemistry calculations. In other words, it provides the simulator with fundamental organic chemistry reactions and calculations to allow for the virtual manipulation interaction of the drug molecule. The London dG scoring function is a mathematical function that scores binding affinity between two molecules. Based on how the Merck molecular force field determines the predicted interaction of drug molecule of the drug molecule and the target molecule (that the drug binds to) to be, the London dG scoring function provides a score based on how effective each interaction is. To reiterate, the London dG scoring function essentially provides a score denoting the projecting efficiency of the interaction between each drug molecule with its target molecule (that the drug binds to). The Merck molecular force field provided the possible orientations of each amixcile and NTZ binding to PFOR and the London dG scoring function scored the free energy of each possible orientation for each amixicile and NTZ in the PFOR active site through these specific calculations. Using the normal interaction of pyruvate and PFOR active site, where pyruvate forms a hydrogen bond with TPP, the Merck molecular force field was used to dock the nitro group of NTZ (the red structure in Figure 2-B) was into the non-reactive PFOR active site in a similar manner. Onto the nitro group of NTZ, the rest of amixicile’s components were added using the Merck molecular force field (head group, amide linker and benzene ring) and different conformations of amixicile were virtually generated. The confirmations with the highest binding affinity were implemented again in the PFOR active site, this time only allowing the amino acids and ligands in the active site to virtually interact with the amixicile. Each of these confirmations were scored via the London dG scoring function (scores based on predicted effectiveness of interaction/binding affinity). The amixicile confirmation with the highest score was then used to further assess and modify the scaffold of amixicile. Specifically, different benzene linkers and tail groups were virtually assessed and scored (via the London dG scoring function) and the orientation with the highest score was chosen as the amixicile model.
|
Result
The simulation predicts that the space for interaction in the PFOR active site may be more abundant with amixicile, compared to NTZ . Specifically, it was observed that the addition of a propylamine tail in amixicile allowed for more interaction with the active site of PFOR. It was virtually predicted that different groups of amixicile interacted with certain conserved amino acid residues in the active site (Figure 3): ArgB114 and ThrB31 with the nitro group (indicated by the blue arrow), AsnB996 with keto group of the amide bond (indicated by the green arrow), and AspB456 with amine of the propylamine tail (indicated by the purple arrow). The binding efficiency of amixicile can possibly be explained by the particular binding spots that MOE predicted. As stated before, MOE predicted that the propylamine tail allowed for tighter binding with the active site. What could be the reason behind this? In figure 3-A, the red spheres indicate the amino acid residue aspartate. The propylamine tail is predicted to bind to this aspartate residue. Since the propylamine tail ends with a positively charged ammonia and the aspartate residue is negatively charged, this may explain why the propylamine tail is projected to bind with the aspartate residue. Spatially, the aspartate residue is a bit further from the other amino acid residue - likewise, the propylamine tail is the furthest extension of amixicile. This is probably why the propylamine tail allows for more tighter binding with the active site. Furthermore the keto group (carbon-oxygen double bond), is projected to bind with the asparagine residue (figure 3A, green residue). As the amine groups in asparagine residue have an unpaired electron and the keto group has an affinity for electrons, this may explain the projected binding between the keto group of amixicile and the asparagine residue. Moreover, the nitro group of amixicile is projected to bind with arginine and threonine residues (fig 3A, orange and yellow respectively). These two amino acid residues are the closest to TPP, which indicates that the nitro group may be involved abstracting a proton from TPP. As explained in the introduction, NTZ (and also amixicile) inactivate TPP by abstracting a proton from TPP. Since the nitro group has a negative charge and the arginine residue is known to have a positive charge, this may partially explain why the nitro group binds to arginine. The threonine residue is uncharged and polar, so the nitro group can also bind to the threonine residue - it may not be as strong of a bond as that with arginine. |
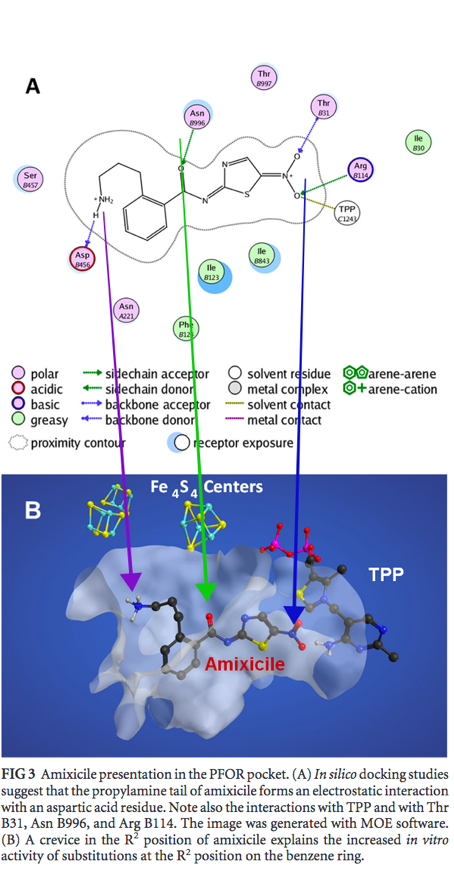
Fig. 3A:Predicted amino residues of PFOR to which amixicile binds [Kennedy et al.].
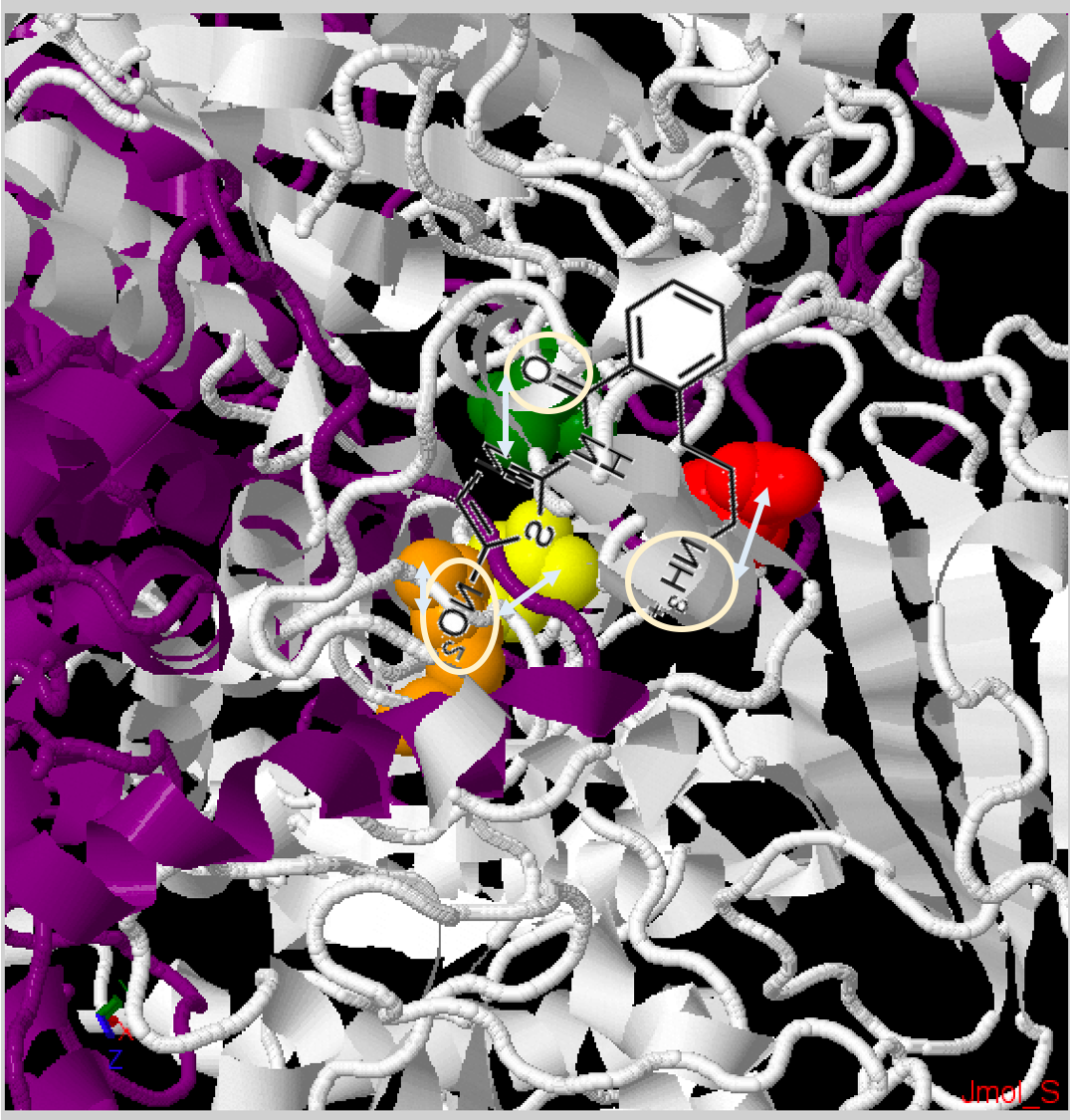
Fig. 3A:Fig. 3B: Zoomed in PFOR structure of Desulfovibrio africanus, particularly the active site. Chain A - Dark Purple, Chain B - White. Squared in white are the amino residues that the simulation predicts interact with amixicile: ThrB31 - yellow, ArgB114 - orange, AsnB996 - green, AspB456 - red. [Source: Jmol Protein Explorer].
|