|
A couple of weeks ago you had the idea of studying Fragile X Syndrome in Drosophila. The plan was to use the known human FMRP protein and then use it to scan the Drosophila genome to find a similar gene. Then it's just a matter of mutating the gene in Drosophila, and investigating what effect such a mutation may have on fly mental function. Then you got stuck. No Drosophila genome means nothing to scan. So for the past two weeks you have been sequencing the Drosophila genome. Now that's behind you, and you have a genome sequence in hand. The time has come to continue with the plan. 1. From the course web site, click on Resources and Links and then on NCBI (National Center for Biotechnology Information, a repository of vast amounts of information).
2.
Click on the down arrow to expand the choices for
the Search box from All Databases, and select Protein.
3. Enter FMRP into the white box, and press Search (or press Enter).
4.
You might expect to pull up an entry for the human
FMRP gene, but no such luck. The search returned a few 100 entries (!), some
from flies – which is interesting (You'll probably have to scroll
down to see them). Let's
try to reduce the number to something more manageable. 5. Click the Advanced Search tab. Type FMRP into a white box next to All fields. To limit the search to the human genome, go to the second All fields box, click the down
arrow to expand the choices from All fields, and select
Organism, then type human into
the box to the right. Finally, choose AND (it's probably already specified) to specify that you're looking for entries that contain both the word FMRP AND
the organism human, click Search. 6. I got about 60 entries now. Certainly an improvement, but
surely humans don't have 60 different FMRP proteins! Part of the problem is that
we're searching multiple overlapping databases and getting the same protein
back multiple times. To cure that, go to the list of Source Databases on the left
side of the screen and click a single database (I
chose GenBank).
7.
Now I'm down to 16 entries, but most don't look very
interesting. The remaining are different from each other in ways that need not concern
us now. Click the entry with the accession number AAH86957.
8. Lots of information here. One important item (scroll down a bit) is a
reference to a journal article that describes the work that led to the
results you're looking at. Clicking on the PubMed number leads you to an
abstract and a link to the full length article -- potentially very useful! Remember
how to do this! But what we're after right
now is the protein sequence, seen at the bottom of the page. The sequence is
given according to the one-letter amino acid codes (see course web site, Links and Resources,
Genetic Code, for a list of them). SQ1. Is this the right sequence? Scrolling back to the top of the page you'll see something labeled 'ORGANISM' and a list of terms starting with "Eukaryota". I'm willing to admit I'm a eukaryote. Chordata, Craniata, Vertebrata... yes, I have a backbone... but am I also a Haplorrhini and a Catarrhini? What do they mean? 9. The display is good for some purposes -- the numbering makes it easy for humans to find what amino acid is at a specific positions -- but computers prefer straight sequence, without numbers or spaces. To get this, scroll back up to the top of the page. You'll see near the top a menu under the Display Settings button, and there you'll find GenPept checked. The format you're looking at is called GenPept (for "GENbank PEPTids"). Change the format to FastA by clicking he down arrow to open a menu and then clicking the button next to FastA and then clicking Apply.
10.
You may already know what FastA
format is, but in any event, if you've gotten this far, you're looking at it: One line
of documentation preceded by ">" and multiple lines of sequence.
If you want to save this file, don't use the browser SAVE function, or you'll get a mess of html browser goo. Instead, click Send to, then click the File radio button. arrow, and finally click Create File to save the file to an appropriate directory on your computer. For now, just copy the sequence, with or without the
documentation line. (In passing, you could also have obtained a savable file by clicking FastA (text) in the previous step) 11. Now we're ready
to do the search. You can initiate the search using the sequence your looking at immediately by clicking Run BLAST in the right column, but for reasons that will become apparent, take a longer route to the same end. Open up the NCBI
icon at the top of the screen in a new tab (on a PC, right click the icon) to return to the NCBI home page, and click BLAST in the Popular Resources
box at the right of the page. BLAST (Basic Local Alignment Search Tool)
is undoubtedly the most widely used bioinformatic tool in existence. We'll
talk about what it does and how it does it, but for now, let's use it to find
the fruit-fly gene we want. We have a protein sequence, and we want to find a
fruit-fly protein, so look under Basic BLAST and click
protein blast.
12. Paste the FMRP
sequence into the Search
box, where you're invited to enter a FASTA sequence (or upload the file, if you saved it) (or type in the accession number), and click the Blast
button near the bottom of the page.
You'll probably get a page pretty quickly
showing conserved domains in the protein -- take a look at them and reconsider SQ2...
More on this another day. For now, wait a bit more to get the full
search results. These results may take tens of seconds to come up or more or
less, depending on the time of day and alignment of the stars. SQ4. (While you're waiting...) What do you think Blast doing? 13. Again, way too many hits! Each red line in the Graphic Summary Section is a very good hit. SQ5. About those thick red lines... What happens when you mouse over one of them? Each line in the Descriptions section gives a link to one of those hits. We'll cut them down in a moment, but first,, scroll down until you reach the Alignments section, which should look something like:
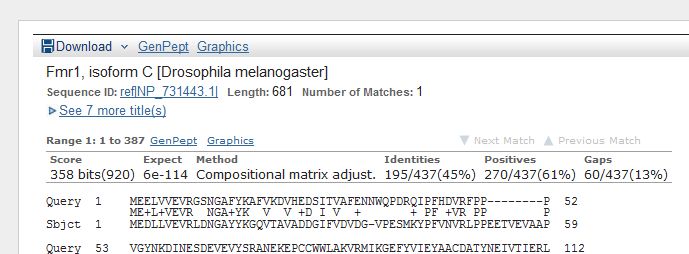
This shows the
alignment of the protein we submitted (human FMR1 protein), i.e., the Query, to the
protein that Blast found, the Subject. The letters to the right look like an amino
acid sequence (check the query sequence -- it should be identical to the sequence you
copied). The sequence to the right of "Sbjct" is the amino acid sequence of a similar protein
found by Blast. The line in between show all the amino acids in common. Looks like the
similarity is pretty good! Indeed, if you look at the Identities statistic above the
sequence, you'll find that the sequences are 100% identical. The best hit happens to be
to the FMR1 protein of Homo sapiens. This should come as no surprise. The protein
most similar to FMR1 is FMR1 itself!
SQ6. Take a look at the best match in Pan troglodytes (chimpanzee), which will be XP_003317789. How many differences are there between the human and chimp proteins? (Examine the statistics at the top of the match) Where is the difference and what is the amino acid change? You'll have to peer very closely at the middle line, between the query sequence and subject sequence, for a position that does not contain a letter. SQ7. Take a look at the best match in Myotis lucifugus (what is that?). How many conservative changes are there? (compare the number of positives to the number of identities). These will be represented by '+' in the middle line. How many nonconservative changes are there? These will be represented by ' ' in the middle line. How do conservative and nonconservative changes differ from one another? Why are these specific amino acid alignments considered conservative or nonconservative? Any other changes besides these? SQ8. What sense can you make of the organisms associated with the hits? Why these and not others?
14.
Unfortunately, none of the matches shown come from flies. Let's help the process.
Click Edit and Resubmit at the top of the page to go back to the Blast submission page (where you pasted in the
protein sequence). You'll see at in the middle of the page a section called Choose
Search Set. In the box marked organism, start typing Drosophila, and click Drosophila
melanogaster. Then click the Blast button again.
15. In several seconds... much better!
Now you have only a few hits, (scroll down) all from Drosophila (of course). Here's the top hit: SQ9. How good is this hit? What's the percent identical amino acids with respect to the human FMR1 protein you used as the query? Does this number correspond to your visual perception of the number of identical amino acids? SQ10. There doesn't seem to be nearly as many identical amino acids as in the previous case. How likely do you think it is that such a match might have arisen by chance? Could this be some random protein that happens to be a little bit similar? There are several entries that have the identical sequence, from three different databases (emb=European Molecular Biology Laboratory; ref=RefSeq; and gb=GenBank). You'll notice that the degree of similarity between the human and fly FMRP protein is much less than between human and mammalian proteins. Only 45% of the amino acids are identical, according to the statistics above the alignment. Is this degree of significant? The Expect value (close to 3e-115... this changes over time) gives you an indication. It says that if you search a database with the size and amino acid composition similar to the one you actually searched but with the sequences randomized, then a match this good or better would arise with a probability of 3 in 10^115 (1 with 115 zeros after it!). In other words, it couldn't possibly have arisen purely by chance in the lifetime of the universe. We'll return later to Expect values, since it is very important to understand what they mean and what they don't mean. By the way, since the GenBank database grows with time, the E-value also changes, so it may not be the number I gave. 16. OK, we've got the protein. But that's not good enough. You can't clone a protein. We need the gene to clone, mutate, and then put it into a fly to make a mutant fly that we can test for physiological function. Click the GenPept link, right above the statistics for the match. This should get you to the protein sequence (verify this by scrolling down to the bottom of the page -- do you see an amino acid sequence or a nucleotide sequence?). To find the gene sequence, look at the DBSOURCE (database source) field near the top of the page. A protein sequence is almost never obtained by direct sequencing of the protein but rather by computer translation of the nucleotide sequence. The DBSOURCE is given as NM_169324.2. Click that link.
17. This brings you
to a page called "Drosophila melanogaster FMR1... mRNA".
The source of the protein sequence was virtual
translation of a cloned piece of mRNA (since you can't clone RNA directly, it
was first made into a DNA copy, or cDNA).
Scroll down to the field called CDS
(CoDing Sequence -- don't ask me why anyone would think this is a proper
abbreviation!). It says that the coding sequence (some would say gene)
extends from position 569 to position 2614 in the cDNA sequence.
18. This may seem a
roundabout path to get to the gene. Why not start with the human gene
(instead of the human protein)
and use it to pick out the fly gene? OK, we'll do it that way. Go back to the NCBI home page.
Make sure Nucleotide is specified in the box at the top of the page. Enter FMRP as
the search term, click Search, and, as you did in Steps 3 through 5, confine the search
to humans and to GenBank and limit the search
to GenBank.
19. Choose the
first complete sequence that encodes FMRP (recalling what "FMRP" stands for) -- it will say "complete cds" in the descriptor line -- and is long enough to contain the sequence you found earlier. Avoid FRMP
interacting protein, which is different from FMRP itself. Get the
sequence from it as before.
20. Find your way to Blast, repeating Step 11, but this time
choose nucleotide blast, using a DNA query to search a database of DNA sequences.
Again paste the sequence into
the window. (This time you may or may not need to change the defaults, specifying that the database is not the
human genome but rather Others). Under Program Selection, Optimize for,
choose "Somewhat similar sequences". Now click the Blast button. When the
results are returned (maybe a minute), you'll see that there are
tons of hits as before, but this time there are many mammalian sequences with multiple
differences with respect to the human sequence. But never mind that -- we're
after flies. So go back to the submission page and specify Drosophila melanogaster and Blast again.
Why such a difference between the protein and nucleotide blasts?
One last trick. Get back to the BLAST front page, either starting over at NCBI or using the browser back button several times. This time choose BLASTX, which takes your nucleotide sequence and translates in all six possible reading frames before comparing the translated sequences against all known proteins. Paste in the human FMRP nucleotide sequence, choose Drosophila melanogaster as the target organism and go. (This might take about six times as long as your previous Blast)
|